Synopsis by Liron Bendor
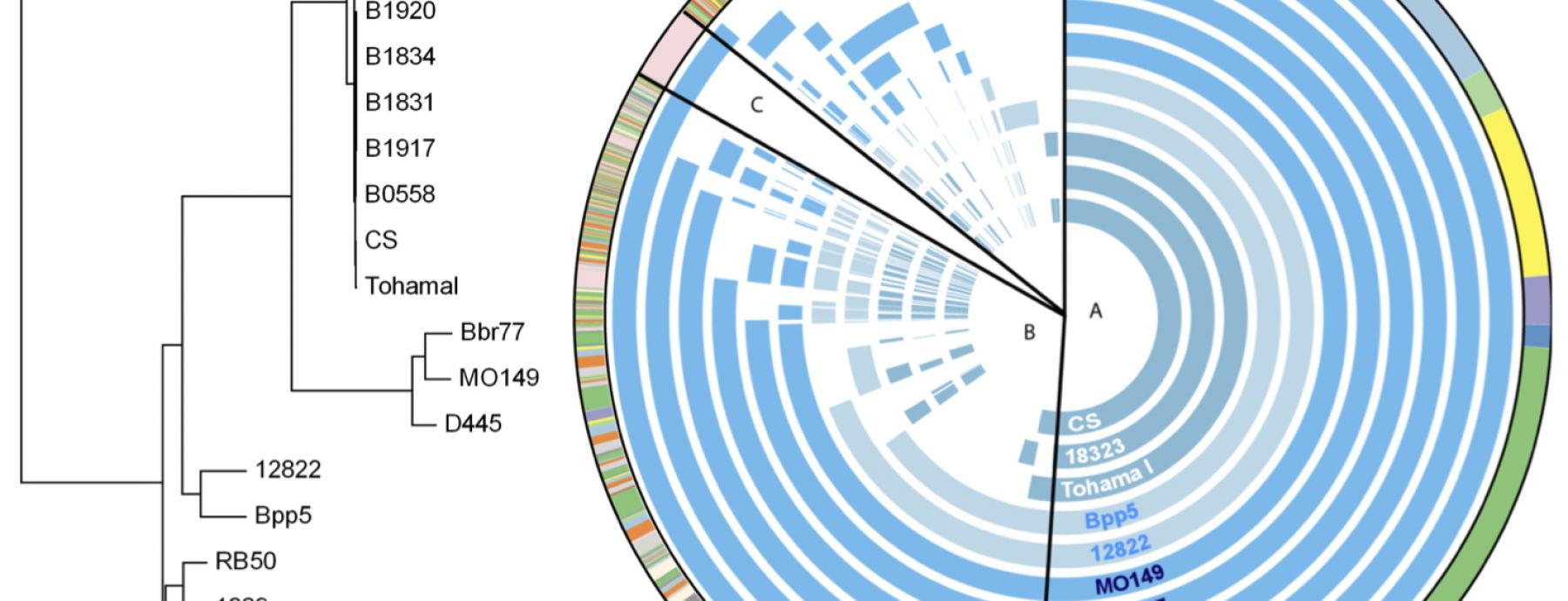
The genus Bordetella contains nine species, three subspecies of which are called “the classical bordetellae.” Despite being closely related, the classical bordetellae differ dramatically in their host range, virulence during infection, and persistence. Bordetella bronchiseptica, which infects mainly non-human mammals, is thought to be the evolutionary progenitor of B. pertussis and B. parapertussis, the etiological agents of whooping cough in humans.
In a study led by CIDD Professor Eric Harvill and published in BMC Genomics, researchers used comparative genomic analyses to reveal relationships between the different classical bordetellae. They sequenced seven classical Bordetella isolates, including five non-human and human-associated B. bronchiseptica isolates, one B. parapertussis strain isolated from sheep, and one B. pertussis strain isolated from humans. Using a genome-wide single nucleotide polymorphism (SNP) analysis on these strains and comparing them to four previously sequenced Bordetella strains, they determined that the human-isolated B. bronchiseptica strains are more closely related to B. pertussis than to B. bronchiseptica strains isolated from non-humans. They also determined that, in contrast to conclusions drawn from multilocus sequence typing (MLST) data in the literature, the sheep-adapted B. parapertussis strain is more closely related to the human-adapted B. parapertussis strain than to non-human B. bronchiseptica strains.
The researchers also assembled the bordetellae pan-genome. In a clear reflection of the diversity of host ranges and virulence characteristics demonstrated by these bacteria, only 50% of the pan-genome was found to be conserved in all strains. The pan-genome also appears to be “open,” or able to acquire new genes; the classical bordetellae may have acquired virulence factors, including O antigen and pertussis toxin, via horizontal gene transfer from yet unknown sources.
This research, beyond revealing the relationships between the classical bordetellae, helps to shed light on potential mechanisms utilized by the bacteria to generate phenotypic diversity and adapt to their hosts.
Written By: Park J, Zhang Y, Buboltz AM, Zhang X, Schuster SC, Ahuja U, Liu M, Miller JF, Sebaihia M, Bentley SD, Parkhill J, & Harvill ET
Paper Url: http://www.biomedcentral.com/1471-2164/13/545/
Journal: 0.920138889
Journal Reference: 0.920138889
Paper Id: 10.1186/1471-2164-13-545